
1:2 - Why Beta Cells Stop Regenerating
- Bowie Matteson
- May 15
- 6 min read

As we saw in Part 1, the question of whether those with T1D have beta cells is not specific enough. We have assumed for decades that T1D is synonymous with beta cell death. We now know that a lack of insulin does not necessarily mean beta cell death. It may be a case of lost identity, a broken or suppressed communication system, or even a prolonged state of dormancy/recovery.
If some beta cells remain—quiet, dormant, dedifferentiated, or structurally altered—the next question is obvious:
Why don’t they simply regenerate?
Why doesn’t the pancreas just replace what’s been lost?
After all, other tissues do. Skin heals. The gut turns over rapidly. The liver can recover from astonishing levels of injury. So why do beta cells seem so reluctant to re-enter the cycle of repair?
The answer, as far as current research can tell, is not that beta cells are incapable of growth. It is that in adulthood they are held behind a series of well-defined molecular brakes. Human beta cells are not simply "inactive" or "incapable". They are actively restrained from proliferating. (Kulkarni 2012)

The Adult Beta Cell: Built for Function, Not Expansion
In early life, beta cells are much more dynamic. They expand in number during infancy and childhood as the body grows and metabolic demand rises. But by adulthood, human beta cells become strikingly resistant to proliferation. Cell turnover slows dramatically, and the pancreas shifts from a growth-oriented organ to a maintenance-oriented one.
This seems to be a protective adaptation. A beta cell is not just any cell. It is a highly specialized glucose sensor, tightly integrated into an electrical, hormonal, and paracrine network. Insulin is an anabolic protein.
Simply put: Adults are not wired for the same growth experienced in childhood.
Uncontrolled proliferation would risk disrupting that precision. So the mature beta cell shifts from flexibility to stability.
That stability comes at a cost:
Adult beta cells are not just slow to regenerate. They are molecularly programmed not to.
If you're familiar with the history of diabetes research, the idea of the slow-to-regenerate adult beta cell became the basis for the fascination with transplant therapies. The idea being that because adults were fated to slow beta cell growth, they would never achieve an adequate supply on their own.
But consider that we may have overstated the permanence of this stability over flexibility. What if, like in the transition from childhood to adulthood, the beta cell was able to take OFF the brakes the same way they turned them on?
The Brake System
Several signaling pathways repeatedly show up in the literature as central barriers to human beta-cell proliferation. These are not fringe theories. They are among the best-defined reasons we currently have for why beta cells struggle to “express themselves” after injury.
1. DYRK1A — The Cell Cycle Gatekeeper
One of the most important proliferation brakes is DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A). DYRK1A helps maintain adult beta cells in a quiescent state by phosphorylating factors that keep the cell cycle turned off. It prevents nuclear retention of NFAT, a transcription factor needed to activate genes involved in replication. When DYRK1A activity remains high, beta cells stay locked in place. (Shen 2015, Guo 2022, Pucelik 2021)
This is why harmine and related compounds have attracted so much attention: they inhibit DYRK1A and allow NFAT to remain active in the nucleus, which can reopen cell cycle entry in adult human beta cells. But importantly, this does not create endless growth. It removes one major brake.
2. GSK-3β — A Suppressor of Growth Signaling
Another major restraint is GSK-3β (glycogen synthase kinase-3 beta). This kinase interferes with insulin signaling and suppresses the Wnt/β-catenin pathway, one of the core developmental and regenerative pathways in many tissues. When GSK-3β is overactive, beta-cell growth, survival, and differentiation signals are dampened. In experimental models, reducing GSK-3β activity improves beta-cell growth and function. (Feng 2012, Liu 2010, Rulifson 2007)
This matters because it links regeneration to the broader metabolic environment. A stressed, insulin-resistant system is often one in which growth signals are being actively muted.
3. The DREAM Complex — Holding Cells in Quiescence
More recently, researchers identified the DREAM complex as a central regulator of adult human beta-cell quiescence. DREAM is a transcriptional repression complex that helps keep beta cells in a non-dividing state. In other words, it acts like a molecular lock on regeneration-related genes. When the DREAM complex is disrupted experimentally, adult human beta cells become more able to proliferate. (Wang 2022, Gojani 2024, Yousefzadeh 2023)
This finding is important because it gives us a more precise explanation for what “quiescence” really means. These cells are not asleep by accident. They are being actively instructed not to divide.

Beta Cells Don’t Just Need a Green Light — They Need the Brakes Released
This is where regeneration conversations often go wrong.
People tend to ask:
What can stimulate growth?
What turns beta-cell proliferation on?
What’s the regenerative compound?
But in many cases the more important question is:
What is keeping these cells turned off?
Because the adult beta cell doesn’t simply lack stimulation. It sits inside a web of inhibitory signals—cell cycle inhibitors, transcriptional repression, inflammatory stress, metabolic noise, and structural burden. Even if one pathway is stimulated, the others may still be suppressing expression.
This is why so many therapies look promising in isolation but underperform in practice. They may press the accelerator while the parking brake is still on.
Identity Matters Too
There’s another reason beta cells don’t regenerate cleanly: a cell must preserve its identity while it re-enters the cycle.
A mature beta cell is defined by transcription factors such as:
Pdx1
MafA
Nkx6.1
These help maintain beta-cell phenotype, insulin gene transcription, glucose sensing, and mature secretory behavior. Under stress, these factors are often reduced, while progenitor-like programs rise. That means the cell is not just prevented from replicating—it may be losing the very instructions that tell it what kind of cell it is supposed to be. (Gojani 2024, Cui 2024)
So even if a beta cell is coaxed into proliferation, the result may not be a strong, mature beta cell. It may be an unstable, partially dedifferentiated cell unless the broader terrain supports maturation too.
This is why regeneration and maturation cannot be separated.
Human Beta Cells Are Different From Rodent Beta Cells
This point deserves emphasis.
Much of the early optimism around beta-cell regeneration came from rodent studies, where beta cells proliferate more readily. But adult human beta cells are much more resistant to replication. What works in mice often produces only modest or highly conditional effects in people. This has forced the field to become more precise and more humble. (Stewart 2015, Jiang 2018, Vasavada 2025)
It also requires we acknowledge the complexity in the systems involved in diabetes development. Much of the study design in mouse model diabetes involves precise genetic mutations and substances tied to beta cell failure (NOD and STZ mouse lines). These mice "develop" diabetes by experiencing precise elimination of beta cell function. The idea being that the value of research really only exists in finding out how to fix beta cells once they've already failed.
But the timeline of diabetes development has changed, as has the organ and signaling systems involved. T1D in humans likely takes much longer than originally anticipated and involves more variables than any research design could reliably account for. Mouse models, with their targeted and relatively abbreviated onset, fail to mimic the series of systemic compensation patterns seen in human T1D.
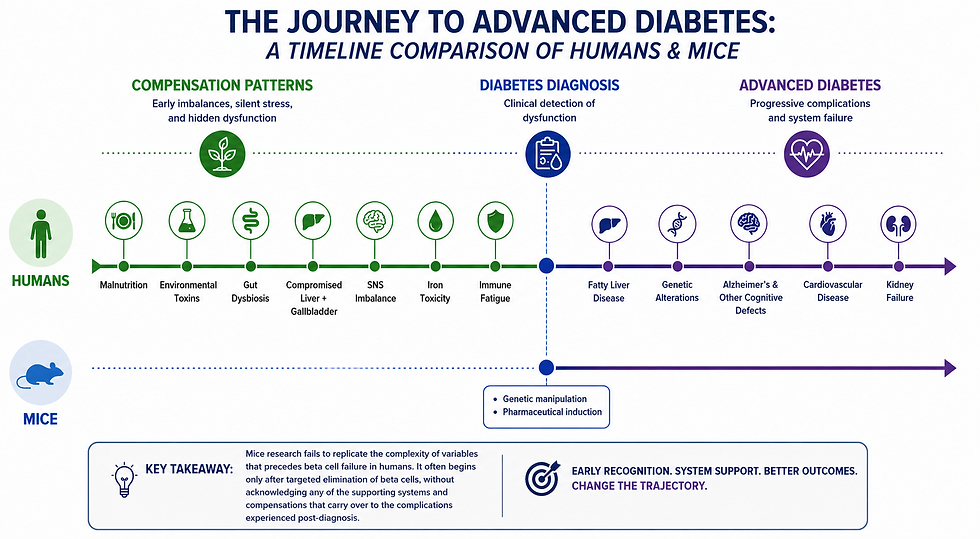
That doesn’t mean regeneration is impossible. It means we need to take human beta-cell biology seriously enough to stop assuming it behaves like rodent tissue.
So Why Don’t Beta Cells “Express Themselves”?
Because expression requires more than survival.
To truly “express themselves,” beta cells need:
permission to re-enter the cell cycle
relief from inhibitory kinases and repression complexes
preservation of identity markers
a metabolic environment that supports growth rather than stress
And in diabetes, many of these conditions are missing at once.
What we call “failure to regenerate” may be less a passive absence of repair and more an active state of suppression.
The beta cell may not be incapable. It may be constrained.
Closing Thought
If the first section asked whether beta cells are actually gone, this section asks a more difficult question:
If some are still there… what is preventing them from returning?
The answer, at least in part, is that adult beta cells are held behind a series of molecular brakes—DYRK1A, GSK-3β, the DREAM complex, identity loss, and the wider stress environment that reinforces them.
Regeneration is not just about pushing growth forward.
It is about understanding what is keeping growth suppressed.
And once those brakes are identified, a new possibility opens up:
Not just preserving what remains—but learning how to let the cells speak again.
Suggested References
Shen, W., et al. (2015). Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Cell Metabolism, 21(6), 1–13.
Wang, P., et al. (2022). Disrupting the DREAM complex enables proliferation of adult human pancreatic β cells. Nature.
Liu, Y., et al. (2010). Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to diabetes. Journal of Biological Chemistry.
Rulifson, I. C., et al. (2007). Wnt signaling regulates pancreatic β cell proliferation. Proceedings of the National Academy of Sciences.
Stewart, A. F., et al. (2015). Human β-cell proliferation and intracellular signaling: Part 3. Diabetes.
Kulkarni, R. N., et al. (2012). Human β-cell proliferation and intracellular signaling. Endocrine Reviews.
Gojani, E. G., et al. (2024). Targeting β-cell plasticity: A promising approach for diabetes therapy. Cells.
Cui, D., et al. (2024). Pancreatic β-cell failure, clinical implications, and therapeutic perspectives. Frontiers in Endocrinology.

Comments