
10 Reasons Viruses Aren't The Reason We Have Diabetes
- Bowie Matteson
- Sep 2, 2025
- 20 min read

Viral takeover is one of the most researched areas of diabetes to date. From Coxsackie B Virus to Epstein-Barre, Covid-19 to the Herpes Simplex Virus, viral infections have been leaving breadcrumbs at the crime scene of diabetes diagnoses as far back as we can remember.

I would argue this correlation stems from the current school of thought on disease development. Acute infection leading to rapid disease onset (AKA The Triggering Event Theory). The idea that a virus has come in and created an environment directly tied to disease outcomes fits that narrative well.
It goes: Our system has been hijacked and manipulated to serve the good of the virus at the expense of our health. Through mimicry, genetic mutation, rewiring signaling patterns and/or flushing the immune system with inflammatory molecules, viruses have a number of documented means of wreaking havoc in a host. Glucose metabolism is the foremost concern of an invading pathogen. Access to energy = higher survivability. In the case of diabetes, a viruses interest in more access to glucose (via higher blood sugars) would make meddling with beta cells a top priority.
Research has found beta cells to be intrinsically hospitable for viruses. Similarly, viruses have adaptations that make them uniquely stealthy in beta cell territory. Things like viral entry receptors that mimic beta cell surface proteins (e.g., CAR, DAF), metabolic vulnerabilities (lax immune tone, increased baseline ROS) and nutrient deposition (high iron) make beta cells an easy and effective target for viruses.
With how that convincing that all sounds, it has become second nature to accept viral influence as a powerful ingredient in diabetes. And this is where the research stops much of the time. The facts fit the theory so the attention moves to the next step. The pathology-driven research turns preventative. Time to focus on the virus. How to alter the receptors, bolster host immunity and silence viral influence becomes the logical next phase.

But what if the theory shifts? We're now seeing deeper and deeper layers of disease development. Traits of cells, tissues, organs and the systems they comprise are showing a longer arc of metabolic collapse than any "triggering event" could explain. There are new variables like nutrient balances and nervous system status. Things once thought to be unquestionable and static traits like receptor presence and immune tolerance turned out to be symptoms of circumstantial adaptations. The proverbial "chicken or the egg?" debate can be applied at any stage of the cell-virus relationship. Are beta cells uniquely vulnerable or have the viruses induced this vulnerable state? Were beta cells always this vulnerable or have they become this way over time given a specific metabolic terrain? If viruses are so tied to diabetes development, why don't more people have diabetes?
When I zoomed out and looked at the trajectory of virus interactions and overlayed my new understanding of the sequences and systems of cell compensation, I didn't see the hostile takeover that virologists and diabetes researchers were selling. There were important questions not being asked. If the cellular terrain has taken longer to become the "pre-diabetic" cell we've come to study in virus interactions, wouldn't we need to peel back the layers of how the cell arrived at this vulnerable state? New timeline, new variables and all?
Because treating the window of transition from no disease to disease as an acute event limited the scope of what research was willing to consider having an influence. Now that the window has been expanded, the number of influences (IE. their timing, sequence and degrees of cellular impacts) grows exponentially.

These questions have lead me to this article and what it includes. I started poking around the pre-existing, pro-virus traits of beta cells and the immune system of those with diabetes. What I found were temporary, reactive and adaptive qualities brought about by external metabolic factors. Factors that we, those with diabetes, could do something about. Out with the helplessness, out with the victimhood and out with trying to outsmart a pathogen that we've willingly let through the front door.
Here's how I approached the topic:
What are the factors with the greatest influence on a successful viral takeover?
What systems are involved in each of the factors?
What specific variables within these systems play into the conditions that precede viral success?
Of all of the systems involved in viral exposure, what are the commonalities across each variable? Do they share common origins?
Are any of those factors inherent to a cell? If not, what is the terrain make-up that leads to these conditions? What's the timeline and sequence of how that happens?
What Are The Top 10 Things That Lead To A Successful Viral Takeover?
🧬 1. Viral Entry Receptors: The Doorway
Every virus needs a point of entry—and this is often dictated by specific cell surface receptors. For instance, Coxsackie B viruses attach to the Coxsackie and Adenovirus Receptor (CAR) and Decay-Accelerating Factor (DAF/CD55) to gain cellular access. These receptors are not static—they are dynamically regulated in response to cellular stress, inflammation, cytokine exposure, and even hormonal signaling. In essence, the presence and abundance of these receptors reflects the current state of the cell, not just a viral targeting strategy. In a healthy cell, these receptors help coordinate immune activity and beta cell coordination. Cells under duress—such as those experiencing endoplasmic reticulum stress, cytokine-induced signaling cascades, or structural remodeling—may upregulate these surface molecules as part of their adaptation or signaling efforts, inadvertently creating more entry points for opportunistic pathogens.
Conventional view: Beta cells express these receptors, allowing viruses direct access and initiating immune targeting. Little attention is given to the circumstances that influence these receptors.
Terrain-first view: Receptor expression is upregulated in response to inflammation, ER stress, and cytokine signaling—all of which may precede infection.
🧪 2. Membrane Fluidity and Lipid Rafts
Cell membranes aren’t homogenous—they contain specialized regions known as lipid rafts, rich in cholesterol, sphingolipids, and signaling proteins. These microdomains serve as essential hubs for cell signaling, immune recognition, and—importantly for viruses—entry and assembly. Viral particles often exploit the structural properties of lipid rafts to dock and enter the cell via endocytosis or membrane fusion. The composition and behavior of lipid rafts are deeply influenced by the cell’s lipid metabolism, which can shift in response to oxidative stress, insulin resistance, or PUFA (polyunsaturated fatty acid) oxidation—particularly when free iron catalyzes peroxidation. Cholesterol synthesis and membrane remodeling often increase under inflammatory stress, which may not be directly caused by a virus, but can make membranes more “permissive” to one.
Conventional view:Viruses exploit these structures to fuse with or enter cells.
Terrain-first view: PUFA peroxidation, chronic inflammation, and cholesterol imbalances (e.g., from liver distress) change membrane dynamics, increasing viral accessibility.
🧲 3. Intracellular Free Iron and Redox Imbalance
Iron is essential for cellular function, but when unbound and poorly regulated, free ferrous iron (Fe²⁺) can act as a dangerous catalyst. Through the Fenton reaction, free iron generates hydroxyl radicals (•OH)—the most reactive and damaging of the reactive oxygen species (ROS). These radicals attack DNA, proteins, and lipids, destabilizing cellular architecture and immune communication. Many viruses rely on high cellular ATP and replication rates, both of which are supported by abundant iron, particularly in the mitochondria. Iron also impairs the function of interferon-stimulated genes, weakening antiviral defenses. However, iron dysregulation may arise independently of viral involvement—through chronic inflammation, poor copper transport, metabolic disease, or disrupted hepcidin signaling. Beta cells have a well documented relationship with iron in both healthy (high metabolic output and mitochondrial density) and diseased states (ROS creation, iron-hiding mechanism, decreased immune surveillance).
Conventional view: Viruses increase ROS to manipulate host machinery and hijack advantageous nutrients.
Terrain-first view: Iron dysregulation precedes infection, undermines antioxidant defenses, and fosters viral replication. Beta cells are iron-rich and mitochondrial-dense. With diabetes, this iron is mismanaged creating a perfect storm of pro-inflammatory conditions with little to no immune defense.
🧬 4. Impaired Interferon Signaling and PRR Function
The first line of antiviral defense involves pattern recognition receptors (PRRs) like RIG-I, MDA5, and TLR3/7, which detect foreign RNA or DNA inside the cell. Once triggered, these sensors initiate a type I interferon cascade, alerting nearby cells and activating immune clearance mechanisms. However, this defense system is vulnerable to both genetic variation and metabolic suppression. Single-nucleotide polymorphisms (SNPs) in genes like IFIH1 (which encodes MDA5) have been linked to impaired viral sensing in T1D. More interestingly, low magnesium levels, glutathione depletion, or oxidative nucleotide damage may impair DNA repair and contribute to mutational burden in these receptors—long before a virus enters the picture. Certain viruses do express proteins to interfere with this pathway, but often they’re exploiting preexisting weaknesses.
Conventional view: Viruses evade or suppress these defenses to remain undetected. This is attributed to the viruses ability to alter host DNA/RNA.
Terrain-first view: PRR polymorphisms may stem from chronic oxidative stress and impaired DNA repair—not solely viral manipulation. The pre-existing pro-inflammatory conditions suppress the body's natural ability to both detect and repair alterations to genetic material.
🗑 5. Suppressed Autophagy and Lysosomal Activity
Autophagy is the process by which cells digest their own damaged components, including organelles, protein aggregates, and intracellular pathogens. It plays a critical role in cellular quality control, energy balance, and immune surveillance. When autophagy is robust, viruses that enter the cell may be captured and degraded before they replicate. However, autophagy depends on tight regulation by AMPK (activated by low energy states) and mTOR (suppressed during fasting or stress). In nutrient-overloaded or sedentary states, chronic mTOR activation inhibits autophagy, leaving cells vulnerable to persistent infections. Some viruses—like Coxsackie B—actively manipulate this process to form autophagosome “replication factories”, but only if the surveillance system is already compromised.
Conventional view: Viruses block autophagy to avoid destruction and create replication niches.
Terrain-first view: Suppressed AMPK, overactive mTOR, nutrient overload, and low cellular energy already compromise autophagy before infection. This creates the perfect environment for viruses to thrive.
🧵 6. ER Stress and the Unfolded Protein Response (UPR)
The endoplasmic reticulum (ER) is the cell’s protein-folding factory. When overwhelmed by excessive protein production, oxidative stress, or calcium imbalance, the ER activates the unfolded protein response (UPR) to restore balance. This includes slowing down protein translation, increasing chaperone production, and—if unresolved—triggering apoptosis. In insulin-producing beta cells, which naturally handle heavy protein traffic, ER stress is common—especially under metabolic or inflammatory pressure. Certain viruses exacerbate ER stress to induce host cell death or delay immune responses, but it’s often preexisting stress that invites them in. Nutrient excess, iron overload, glutathione depletion, and persistent inflammation all tip the balance toward ER dysfunction before infection occurs.
Conventional view: Viruses increase ER stress to create conditions for replication and immune disruption.
Terrain-first view: Beta cells naturally endure high ER load, and nutrient imbalances, redox instability, and GSH depletion increase stress well before viral engagement.
🧬 7. Epigenetic Permissiveness and Chromatin Relaxation
The structure of chromatin—whether tightly packed or loosely unwound—determines which genes are accessible to transcription. Viruses rely on host transcription machinery to replicate, and so they favor a relaxed chromatin state. This “permissiveness” arises from hypomethylation, histone acetylation, and weakened epigenetic control. But what opens the chromatin in the first place? Folate, B12, choline, zinc, and S-adenosylmethionine (SAMe) are all required for DNA methylation. Deficiencies in these nutrients, along with chronic inflammation or environmental toxins, can lead to a global loss of epigenetic precision. Certain viruses can manipulate chromatin structure to maintain latency (like EBV), but the groundwork is often laid by terrain-level instability.
Conventional view: Viruses manipulate host epigenetics to enable latency and replication.
Terrain-first view: Folate/B12 deficiency, chronic inflammation, and oxidative stress cause hypomethylation before viral arrival.
⚡️ 8. Mitochondrial Dysfunction and Bioenergetic Collapse
Mitochondria are not just the powerhouses of the cell—they’re critical for redox signaling, apoptosis regulation, and immune activation. A healthy mitochondrion buffers ROS, produces ATP, and communicates with the nucleus to regulate gene expression. But when mitochondria are overloaded—by iron accumulation, electron transport chain (ETC) dysfunction, or nutrient excess—they leak ROS and lose membrane potential. This not only destabilizes metabolism, but also weakens antiviral immunity. Some viruses target mitochondria directly, altering fission-fusion dynamics or triggering mitophagy. But most viral manipulation occurs after mitochondrial decline is already in motion. In T1D, beta cells often exhibit mitochondrial fragility long before a virus is detected.
Conventional view: Viruses damage mitochondria to disable antiviral defenses.
Terrain-first view: Iron overload, nutrient deficiencies, and poor circadian rhythm precede infection and impair mitochondrial function.
🛡 9. Immune Privilege and Inflammatory Silence
Certain tissues—like the brain, eyes, and pancreatic islets—are considered immune-privileged, meaning they are typically not actively surveilled by immune cells unless damaged. This helps prevent autoimmune overactivation in delicate structures. But it also means that cells in these areas, like beta cells, can suffer internal dysfunction for long periods without triggering an immune response. Only when damage-associated molecular patterns (DAMPs) are released—such as from mitochondrial breakdown or ER rupture—do immune cells begin to take notice. This explains how a virus might seem like the "first responder," when in fact it’s reacting to the unresolved metabolic fire already smoldering.
Conventional view: Viruses attack immune-silent beta cells, triggering an immune reaction that initiates T1D.
Terrain-first view: Beta cells only become visible after metabolic distress signals or cell death alert the immune system. Viruses are opportunistic in cells exhibiting advanced signs of decline.
🧹 10. Proteasome Function and Antigen Presentation
The proteasome breaks down damaged or misfolded proteins into peptides for recycling or immune presentation via MHC class I molecules. This process is central to flagging infected cells for immune clearance. Some viruses (e.g., EBV) produce proteins like EBNA-1 that resist proteasomal degradation, helping them avoid detection. But proteasome function is also impaired by oxidative damage, AGE formation, and inflammatory lipid byproducts. Cells under chronic stress may already show signs of antigen presentation failure before viral interference begins. Thus, viral immune evasion is often a symptom of deeper dysfunction in protein processing and cellular waste management.
Conventional view: Viruses evade degradation using specialized proteins (e.g., EBV’s EBNA-1).
Terrain-first view: Proteasome dysfunction is common in oxidatively stressed, nutrient-deficient, or aging cells—before viral engagement.
There is an undeniable underlying issue in each and every one of the factors influencing viral success that is completely independent of the specific viral ability. We've approached the viral origin theory of diabetes without considering the state of the cell beforehand. It's clear to see that metabolically, structurally and nutritionally speaking, beta cells have gone through a long arc of collapse and compensation. Viruses are not the source of the disruption, they are opportunistic scavengers.
Viruses are like hackers:
They don’t build the broken system.
They don’t install the backdoors.
But once a system is overloaded, misconfigured, or unpatched, the hacker gets in easily—and might even look like the main problem if you're not reading the logs correctly.
So:
Is the virus the initiator? In rare acute scenarios—maybe.
In chronic, low-grade terrain deterioration? No. The terrain falls apart first.
Viruses are opportunists. The real question is: what gave them the opportunity?
1. Viral Entry Receptors: Upregulation as a Symptom, Not a Cause
Root causes: ER stress, oxidative stress, cytokine exposure, and even exposure to advanced glycation end-products (AGEs) upregulate entry receptors.
Nutrient drivers: Excess glucose, iron, or omega-6 PUFAs → ROS → NF-κB activation → transcription of adhesion molecules.
Conclusion: The metabolic and inflammatory state of the cell creates the door; the virus simply walks through it.
2. Membrane Fluidity and Lipid Rafts: Cholesterol & PUFA Reactions as Terrain Signals
PUFA peroxidation, driven by Fe²⁺ and mitochondrial ROS, destabilizes lipid rafts.
Hepatic cholesterol output, typically cast as the villain, may be a protective compensatory response to inflammatory stress.
Inflammation → SREBP activation → cholesterol synthesis: An attempt to stabilize membranes, not random chaos.
Conclusion: The membrane vulnerability is an adaptive response gone awry, and viruses merely exploit that opening.
3. Free Iron and Redox Vulnerability: The Primordial Disruptor
Excess labile iron doesn’t just make the terrain hospitable to viral replication—it creates the conditions for transcriptional chaos, mitochondrial failure, and even immune dysfunction.
Iron-mediated Fenton reactions predate the virus, set the redox imbalance, and erode the cellular perimeter.
Conclusion: Viruses don’t generate free iron—they capitalize on it.
4. Interferon Signaling & PRR Dysfunction: What Influences Polymorphism?
Polymorphisms don’t arise in a vacuum. Impaired DNA repair mechanisms, epigenetic instability, and oxidative nucleotide damage drive mutations that compromise pattern recognition receptors (PRRs).
Nutrient co-factors like magnesium, zinc, selenium, B12, and folate all impact:
DNA repair
Methylation capacity
Histone maintenance
Conclusion: PRR dysfunction might be exacerbated by viral mechanisms, but it is primed by intracellular insufficiencies and environmental exposures.
5. Suppressed Autophagy & Lysosomal Activity: A Terrain Failure First
mTOR overactivation (via excess amino acids, insulin, or caloric abundance) suppresses autophagy.
Low AMPK (due to sedentary behavior, nutrient excess, or metabolic inflexibility) further prevents autophagy initiation.
Iron overload also suppresses lysosomal acidification, interfering with cleanup mechanisms.
Conclusion: Autophagy is already failing in a stressed cell. The virus takes advantage, but doesn’t necessarily create this terrain.
6. ER Stress and the UPR: Cause or Effect?
ER stress can be worsened by viral activity, but it’s often preceding viral entry in metabolically burdened cells.
Excess protein load, disulfide bond misfolding, and glutathione depletion impair ER function.
Chronic unfolded protein response (UPR) → inflammation → receptor upregulation → increased viral susceptibility.
Conclusion: Viruses weaponize a failing ER—they don’t originate its breakdown.
7. Epigenetic Permissiveness: Who’s Unlatching the DNA?
You’re right to ask what causes hypomethylation.
Drivers of low DNA methylation:
Folate / B12 / choline deficiency
Chronic inflammation (TNF-α, IL-6 reduce methyltransferase activity)
Oxidative damage to methyl donors (via iron, homocysteine)
EBV and others exploit this, but it's terrain first.
Conclusion: A destabilized epigenome is the key—viruses are the lockpick.
8. Mitochondrial Dysfunction: Weak Before the War
Viruses prefer metabolically active cells, but beta cells often already show signs of:
ETC leakage
Reduced ATP output
Glutathione depletion
Excess superoxide
Compounding causes: Iron overload, low CoQ10, magnesium deficiency, chronic hyperglycemia, or amino acid imbalance.
Conclusion: Mitochondria are faltering before the virus shows up—leaving cells wide open to manipulation.
9. Immune Privilege: Islet Isolation and the Myth of the Healthy Target
Beta cells aren’t inflamed until damage accumulates.
So what sparks the inflammation?
If we acknowledge the above terrain breakdowns…
Then viruses might not "invade" healthy beta cells—they may only gain traction after years of unresolved intracellular stress.
Conclusion: Viral targeting of healthy beta cells is unlikely. Instead, they exploit cells already signaling distress.
10. Proteasome Dysfunction: EBNA-1 Is Effective, But Not Alone
EBV’s EBNA-1 avoids degradation, but:
Chronic ROS exposure impairs proteasomal activity.
AGEs and lipid peroxides modify proteins, jamming the system.
Nutrient sufficiency (especially magnesium, selenium, cysteine) is required for proper function.
Conclusion: The proteasome is often already inefficient in inflamed or metabolically stressed cells. EBV’s tactic is opportunistic—not initiating.
Once we start to see the virus as a symptom of a compromised system, they lose their power. A healthy system has the immune capabilities, the DNA protection and repair, and the resource conservation to handle viral exposure. Viruses are not new to humans. I would argue they are not outcompeting us in our co-evolution. We simply just aren't tending to our body's fundamental requirements. Vitamins, minerals, amino acids, fatty acids and energy sources that maintain our innate defenses. We have become lost in seeking to alter something outside of ourselves at the expense of caring for and tending to our own needs.
The real question then becomes: What is happening in the cell that creates this inflammatory inertia? Is there a single source of dysfunction that becomes an energy sink in the body's self-corrective abilities?
My guess is no. What we're experiencing in the current healthscape extends back far beyond our lifetimes. Epigenetically speaking, we've been inheriting imbalanced and dysregulated nutrient and hormonal profiles with their respective compensation patterns for generations. Each generation compounds the previous. Add in the steadily depleted nutritional status of the foods we eat, the symptom-management origins of the medicine we rely on and the pro-inflammatory lifestyles our cultures promote, and you've got a million different reasons our cells are screaming bloody murder.
That being said, not all is doom and gloom. I want you all to leave this article with a faith in your body's ability to self-correct. Let's look back to the questions we began with:
Of all of the systems involved in viral exposure, what are the commonalities across each variable?
Do they share common origins?
Are any of those factors inherent to a cell? If not, what is the terrain make-up that leads to these conditions?
What's the timeline and sequence of how that happens?
🔎 Investigating the Deeper Pattern: What Opens the Door for Viral Success?
1. What are the commonalities across each variable?
Across all 10 viral susceptibility factors, we find several recurring themes:
✅ Loss of Barrier Integrity
Membrane fluidity, tight junction breakdown, and epigenetic relaxation all reflect a loss of control over what gets in and out.
Whether it’s the lipid membrane, DNA structure, or mitochondrial boundary, the theme is clear: the gates are left ajar.
🔥 Chronic Low-Grade Inflammation
Inflammatory signaling shows up everywhere: it upregulates viral receptors, disrupts ER function, suppresses interferon signaling, and destabilizes DNA methylation.
Inflammation isn’t always overt—it may simmer for years as a result of poor nutrient status, toxin load, or gut-liver leakage (AKA compromised detox).
⚡ Oxidative Stress and Redox Collapse
From iron-driven ROS to mitochondrial leakage, oxidative stress both damages host structures and impairs immune detection.
ROS directly impact the proteasome, epigenetics, cell signaling, and pattern recognition receptors—a multi-system disruption.
🚨 Nutrient-Linked Surveillance Impairment
Several systems (autophagy, methylation, mitochondrial repair, proteasome function) require:
Magnesium
Zinc
Copper
B12
Choline
Glutathione
Folate
Deficiencies across these nutrients often mirror poor terrain and make immune blindness more likely.
🧬 Compromised Damage Response Systems
The interferon cascade, UPR, and antigen presentation all represent how the cell reacts to distress.
When those systems are weakened, viral detection is delayed or absent.
In summary, these aren't 10 separate problems. They’re 10 different manifestations of terrain collapse.
2. Do they share common origins?
Yes—most can be traced back to a core set of disruptions:
🔗 Iron Overload + Poor Iron Recycling
Iron is the common denominator across ROS, ER stress, immune suppression, and viral replication.
It acts as both a fuel and a sabotage agent, catalyzing damage in DNA, lipids, and proteins.
🧠 Mitochondrial Miscommunication
Mitochondria dictate not just energy, but immune training, apoptotic thresholds, and hormetic responses to stress.
Dysfunction here reverberates throughout the terrain.
This may be a symptom of iron mismanagement
Mitochondria are the most iron-dense organelle (and the most ROS dense...)
🧂 Electrolyte and Mineral Imbalance
Magnesium, zinc, copper, and selenium govern hundreds of intracellular processes.
Chronic depletion limits:
Antioxidant enzyme activity (SOD, GPx)
DNA repair
Energy transfer
Immune function
They all serve as cofactors and co-regulators in proper iron metabolism.
🧬 Impaired Methylation + Epigenetic Drift
Without proper methyl donors (folate, B12, choline), DNA remains hypomethylated, transcription becomes erratic, and immune modulation falters.
This opens the door to both mutation and viral integration.
🌀 Chronic Inflammation and Compensatory Remodeling
The body, in attempting to adapt to stress, may inadvertently upregulate proteins (like DAF or CAR) that viruses exploit.
These changes are not pathogenic by nature—they’re signs of a cell trying to survive in adverse conditions.
3. Are any of these factors inherent to the cell? Or are they terrain-induced?
This is the crux of the terrain-first perspective.
➤ Inherent factors (present in every cell):
Expression of viral entry receptors (e.g., CAR, DAF)
Presence of PRRs and interferon machinery
Proteasome, lysosome, and autophagy pathways
Mitochondria, ER, and redox cycling mechanisms
These are normal tools a cell uses for life, communication, and repair.
➤ What changes is their behavior, which becomes:
Overactive (e.g., chronic UPR or inflammation)
Under-responsive (e.g., suppressed PRRs or autophagy)
Disrupted by nutrient scarcity, toxin exposure, or inflammatory load
Thus, none of the viral susceptibility factors are inherently dysfunctional—they become dysfunctional in the presence of a destabilized terrain.
4. What’s the timeline and sequence of how that happens?
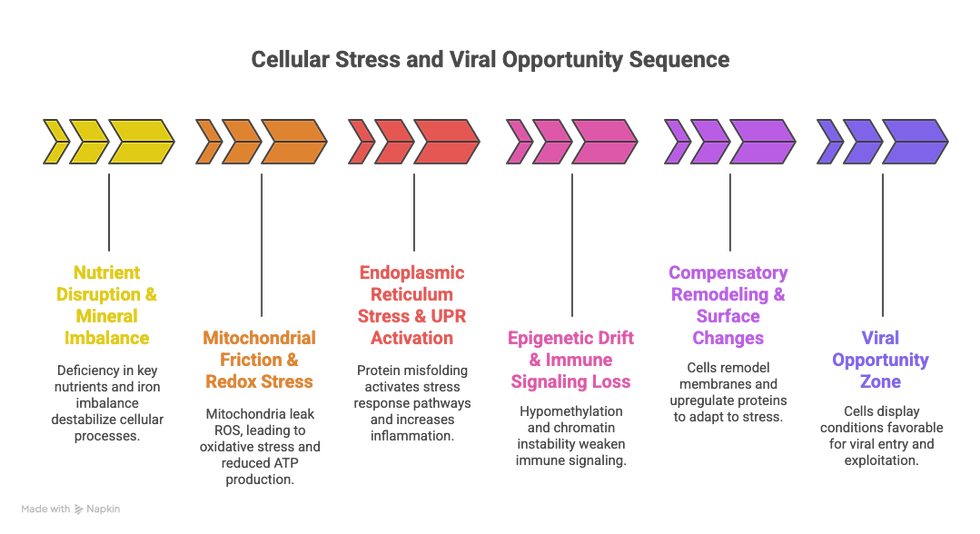
Here’s a possible sequence of cellular breakdown leading to viral vulnerability. It may vary by individual, but this general model offers a compelling terrain-first timeline:
Step 1: Nutrient Disruptions & Mineral Imbalances
↓ Magnesium, zinc, selenium, folate, choline
↑ Iron, copper dysregulation, or excess heme intake
Begins to alter redox signaling and epigenetic stability
Step 2: Mitochondrial Friction + ROS Generation
Mitochondria begin leaking ROS
ATP drops, AMPK is suppressed, autophagy stalls
Glutathione stores are depleted
Redox stress begins to interfere with PRR function, proteasome efficiency, and ER health
Step 3: ER Stress + Unfolded Protein Response
Protein folding errors increase
UPR activation triggers inflammation and cytokine release
Surface proteins (CAR, DAF, ICAM-1) upregulated
Step 4: Epigenetic Drift and Immune Fog
Hypomethylation loosens transcriptional control
Pattern recognition receptors become less effective
IFN signaling is dampened
Step 5: Compensatory Remodeling
Cell attempts to “adapt” by shifting protein expression, membrane composition, or antigen display
Proteasome function lags behind
Lysosomal clearance becomes sluggish
Step 6: Vulnerability Zone
Virus encounters a cell already showing:
Weak immune signaling
Open chromatin
High ATP demands
High iron
Inflammation-induced entry receptors
The virus doesn’t have to “invade”—it’s invited.
🧭 Rewriting the Viral Narrative—From Fear to Foundation
For decades, the dominant narrative around viruses in type 1 diabetes has focused on pathogen as predator. Certain viruses—like Coxsackie B or EBV—were cast as the initial spark, the sole culprits triggering autoimmunity and beta cell destruction.
But as we peel back the cellular layers, a more complex—and empowering—story unfolds.
We’ve explored ten cellular features commonly associated with viral infection:
Receptor expression
Membrane fluidity
Free iron accumulation
Mitochondrial dysfunction
ER stress and UPR
Suppressed autophagy
Epigenetic drift
Impaired immune signaling
Proteasome breakdown
Immune invisibility
While these are often described as targets of viral manipulation, the truth is that they are often the result of terrain-level instability long before viral exposure occurs.
At the heart of each factor, we find a familiar thread:
Nutrient depletion
Oxidative stress
Poor iron handling
Immune disorientation
Structural remodeling as a survival response
These aren’t random weaknesses. They are patterns of adaptation in a system that is trying—desperately—to maintain function under pressure.
The virus, then, is not the architect of destruction. It is the opportunist that enters after the blueprint has already begun to unravel.
🔄 From Vulnerability to Resilience
When we understand this sequence, the paradigm shifts:
Viral presence no longer equals viral causation.
Immune activation is no longer an irrational betrayal—it’s a rational response to cellular distress.
And perhaps most importantly: The power to restore resilience lies within reach.
🌱 So, what now? How do we rebuild the terrain?
Healing doesn't require perfect control. It requires informed stewardship.
✴️ Rebuilding begins by:
Replenishing essential nutrients (magnesium, zinc, choline, selenium, B12, folate)
Balancing iron metabolism, not just lowering intake but improving recycling and transport
Supporting mitochondrial capacity through movement, light, and gentle hormesis
Enhancing redox stability with antioxidant-rich foods, deep breathing, and blood sugar regulation
Reinvigorating autophagy and detoxification via fasting windows, sleep, and herbal allies
Restoring communication between immune and endocrine systems, not suppressing one to “fix” the other
This is not about eliminating viruses. It’s about removing the conditions that make their engagement catastrophic.
💡 A Final Word: Empowerment Over Fear
Understanding that viral involvement is not predestination is perhaps the most powerful takeaway of all.
You are not a hostage to your exposures.
You are not broken because a virus once passed through.
You are not destined for decline because your body was once vulnerable.
Instead, you are being invited—through inquiry, awareness, and grounded action—to rebuild the integrity of your system from the inside out.
Not to fight the virus. But to make the body a place where harmony, not havoc, takes hold.
Support the Channel
📚Buy my eBook documenting my dive into the iron overload-diabetes connection: https://bowie1.gumroad.com/l/ironclad
💊 Interested in the supplements I use?
Thorne Research: https://www.thorne.com/u/bowiematteson
MitoLife: https://www.mitolife.co/discount/BOWIE15
Ancient Bliss: https://www.ancientbliss.com/bowie_matteson
Crucial FOUR: https://www.crucialfour.com/BOWIE15
Microbiome Testing with Customized Probiotics and Supplement Stacks from Viome: http://viomehq.sjv.io/Jz1x1Q
Whole Food, non-GMO Meals from Splendid Spoon: http://slendidspoon.pxf.io/Xm10g5
Additional resources (nutrition guide, supplement guide, exercise) https://bowie1.gumroad.com
📚 Reading Material: Scientific Foundations for the Terrain-Virus Framework
🧬 Viral Entry Receptors (CAR & DAF) in Beta Cells
Bergelson JM, et al. (1997). “Coxsackievirus and adenovirus receptor is a common receptor for Coxsackie B viruses and adenoviruses.” J Virol.https://doi.org/10.1128/JVI.71.10.7801-7807.1997
Stone VM, et al. (2019). “DAF (CD55) upregulation in islets in response to viral stress and inflammation.” Diabetologia.https://doi.org/10.1007/s00125-019-4935-2
🧲 Iron, Oxidative Stress, and Viral Replication
Drakesmith H & Prentice AM. (2012). “Hepcidin and the iron–infection axis.” Science.https://doi.org/10.1126/science.1224577
Gonzalez H, et al. (2018). “Iron metabolism and oxidative stress in viral infections.” Front Cell Infect Microbiol.https://doi.org/10.3389/fcimb.2018.00344
Girelli D, et al. (2021). “The role of iron metabolism in infection and immunity.” Immunity.https://doi.org/10.1016/j.immuni.2021.01.015
🔥 Chronic Inflammation & Viral Susceptibility
Hotamisligil GS. (2006). “Inflammation and metabolic disorders.” Nature.https://doi.org/10.1038/nature05485
Rouse BT & Sehrawat S. (2010). “Immunosuppression during viral infections: roles of inflammatory mediators and cell regulatory pathways.” Nat Rev Immunol.https://doi.org/10.1038/nri2711
🧪 Endoplasmic Reticulum Stress & the UPR in Beta Cells
Engin F. (2016). “ER stress and the UPR in beta-cell biology and diabetes pathogenesis.” Curr Diab Rep.https://doi.org/10.1007/s11892-016-0746-2
Ron D & Walter P. (2007). “Signal integration in the endoplasmic reticulum unfolded protein response.” Nat Rev Mol Cell Biol.https://doi.org/10.1038/nrm2199
⚡ Mitochondrial Dysfunction, ROS, and Terrain Collapse
Newsholme P, et al. (2007). “Mitochondria and beta-cell function.” Diabetes Obes Metab.https://doi.org/10.1111/j.1463-1326.2007.00784.x
Murphy MP. (2009). “How mitochondria produce reactive oxygen species.” Biochem J.https://doi.org/10.1042/BJ20081386
Tiku V, et al. (2020). “Mitochondrial functions in infection and immunity.” Trends Cell Biol.https://doi.org/10.1016/j.tcb.2020.03.007
🔍 Pattern Recognition Receptors (PRRs) and Viral Immunity
Nejentsev S, et al. (2009). “Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes.” Science.https://doi.org/10.1126/science.1167728
Kawai T & Akira S. (2010). “The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors.” Nat Immunol.https://doi.org/10.1038/ni.1863
🗑 Autophagy, Immune Clearance, and Viral Exploitation
Deretic V & Levine B. (2009). “Autophagy, immunity, and microbial adaptations.” Cell Host Microbe.https://doi.org/10.1016/j.chom.2009.08.010
Shi Y, et al. (2013). “Autophagy in beta-cell function and survival.” Autophagy.https://doi.org/10.4161/auto.26170
🧬 Epigenetic Vulnerability and Methylation Drift
Anderson OS, et al. (2012). “Epigenetics: mechanisms, targets, and the role of nutrition in disease susceptibility.” Physiol Rev.https://doi.org/10.1152/physrev.00003.2011
Kuss-Duerkop SK & Westrich JA. (2018). “Viral manipulation of host epigenetic machinery.” Curr Opin Virol.https://doi.org/10.1016/j.coviro.2018.03.002
🛡 Immune Privilege and the Islet Microenvironment
Coppieters KT, et al. (2012). “Imaging of beta-cell destruction in type 1 diabetes.” Cell Metab.https://doi.org/10.1016/j.cmet.2012.06.003
Wekerle H. (2008). “Immune privilege and the control of autoimmunity.” Nat Immunol.https://doi.org/10.1038/ni1008-939
🧹 Proteasome Function & Antigen Presentation
Groettrup M, et al. (2010). “Regulation of antigen processing by the proteasome.” Nat Rev Immunol.https://doi.org/10.1038/nri2773
Levitskaya J, et al. (1995). “Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1.” Nature.https://doi.org/10.1038/375685a0
🧭 Terrain-First Framework & Systems Thinking
Paun A & Danska JS. (2015). “Immunogenetics of type 1 diabetes: model and mechanism.” Trends Immunol.https://doi.org/10.1016/j.it.2015.01.008
Fasano A & Shea-Donohue T. (2005). “Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases.” Nat Clin Pract Gastroenterol Hepatol.https://doi.org/10.1038/ncpgasthep0113



Comments